化学科学与工程学院温鸣团队实现精确定位单原子催化析氢,研究成果发表于《先进功能材料》
来源:化学科学与工程学院
时间:2024-05-08 浏览:
碱性电催化分解水是工业上大规模生产高纯度氢最高效、最环保的方法之一,其中高效经济的电催化剂一直是工业上追求的关键目标。单原子(SA)催化剂由于具有原子效率高、电子结构独特、配位环境易于调节等特点,显示出高活性和低成本的优势,近年来受到了极大的关注。层状双金属氧化物(LDOs)因其二维平面比表面积大、稳定性优异,可以作为贵金属SA催化剂的适宜载体。金属氧化物结合Pt SA催化剂在碱性析氢反应(HER)中虽已表现出巨大的潜力,但由于Pt和载体之间的电子相互作用不足而受到严重限制。同济大学化学科学与工程学院温鸣教授团队积极探索解决方案,利用双离子刻蚀结合原位相变技术,实现了精准定位单原子催化促进析氢反应,揭示了单原子的精确定位对设计高效催化剂的重要性,相关研究成果以“Coupling Interaction Between Precisely Located Pt Single-atoms/clusters and NiCo-Layered Double Oxide to Boost Hydrogen Evolution Reaction”为题,近日在线发表于材料领域著名期刊《先进功能材料》(Advanced Functional Materials)。

研究团队利用双离子刻蚀结合原位相变技术,实现了将Pt SAs和Pt团簇精确定位于NiCo LDO层内及表面(Pt-NiCo LDO),Pt SAs插入到LDO层中通过占据部分Ni原子的位置形成Co-Pt键,促进了Pt与NiCo LDO之间有效的电子相互作用。这些层内Pt SAs与表面Pt团簇协同作用下,HER过程中H2O优先吸附于Pt团簇,其中H*吸附在层内Pt SAs上方的O位点上,显著加速了碱性介质HER过程中H2O的解离,由此获得了比20 wt%的商用Pt/C高近6倍的优异碱性HER性能。
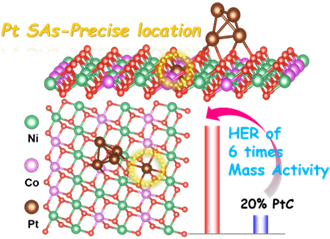
研究人员通过双离子刻蚀结合原位相变技术得到了具有十二面体空心结构和层状纳米片形貌的Pt-NiCo LDO材料。通过球差校正电子显微镜观看到Pt SA(绿色圆圈标记的亮点)和Pt团簇(橙色圆圈),且单原子的总量大于团簇,表明Pt是由单原子和团簇共存的形式存在。X射线吸收谱检测到明显的~1.50 Å Pt-O峰和~2.40 Å Pt-Pt峰,进一步证明了Pt SA和Pt团簇共存于Pt-NiCo LDO材料中。小波变换分析结果显示,在Pt-NiCo LDO中检测到5和7.5 Å-1附近的峰值,对应于Pt-Cl和Pt-M(M=Ni/Co)。因此,Pt原子被插入到LDO层中并与Ni或Co结合,而不是简单地位于层的表面。结合位点吸附的理论计算证实,Pt-NiCo LDO中Pt SA插入NiCo LDO层中是通过占据部分Ni位置形成Pt-Co键,保证了Pt与NiCo LDO之间的强电子相互作用。

研究人员进而对比了不同材料的HER性能,Pt-NiCo LDO在10 mA cm-2时有着92 mV的过电位,Tafel斜率为73 mV dec-1;最佳Pt-NiCo LDO-5.06 wt%催化剂的质量活性几乎是20 wt%商用Pt/C催化剂的6倍,这表明HER中Pt利用率的提高是由于Pt团簇和SA在Pt-NiCo LDO中共存;且Pt-NiCo LDO表现出稳定的电位,在105小时后仅有着50 mV的衰减,证明了其优异的HER稳定性。

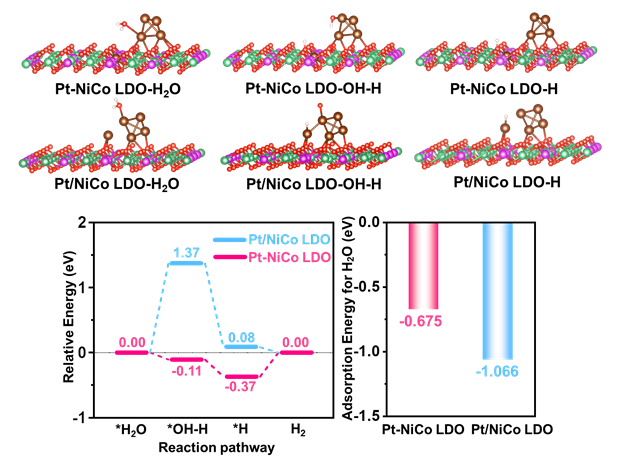
通过理论计算,研究人员探究了精确插入层内的Pt SAs对HER性能的影响及与Pt团簇的相互协同作用。首先H2O被优先吸附到Pt团簇上,由于Pt SA插入到LDO层中,H*优先被吸附到位于Pt SA顶部的O位点上。特别是Pt-NiCo LDO的氢吸附吉布斯自由能(ΔGH)为-0.37eV,而Pt/NiCo LDO为0.08eV,Pt-NiCo LDO中O位对*H的吸附量远大于Pt/NiCo LDO中Pt位*H的吸附量。上述理论结果与实验结果吻合较好,证明了Pt-NiCo LDO在LDO层中插入Pt SAs有利于H2O解离,从而实现了具有高催化活性的电催化剂。
温鸣教授和吴彤研究员为论文共同通讯作者,博士生田亚坤为论文第一作者,Northumbria University傅永庆教授等为研究工作提供了支持。该研究工作得到了国家自然科学基金、上海市科委以及同济大学学科交叉联合攻关项目的资助。
论文链接:https://doi.org/10.1002/adfm.202405919



