针对JAK2-V617F突变型白血病,现有靶向治疗正陷入“疗效短暂”与“耐药加剧”的双重困局。一方面,传统JAK抑制剂无法精准区分突变与野生型蛋白,在抑制肿瘤的同时也“误伤”正常JAK2功能,导致血小板减少等严重副作用,治疗窗口狭窄;另一方面,长期用药后,肿瘤细胞极易通过异常磷酸化、异源二聚化等机制逃避药物抑制,不仅导致疾病复发,还可能快速进展为继发性急性髓系白血病(sAML),患者五年生存率甚至低于10%。
传统药物“只抑不除”的局限,呼唤着新一代疗法的出现——亟需一种能够精准识别突变蛋白并从细胞层面彻底“清除”致癌靶点的治疗策略,以突破耐药瓶颈,实现持续缓解。
12月3日,同济大学生命科学与技术学院杨静教授、武汉大学蒋白山教授、同济大学附属同济医院梁爱斌教授、山东大学赵保兵教授合作在《先进科学》(Advanced Science)上发表题为“Targeting DESI2 as a Novel Therapeutic Strategy for JAK2-Mutant Leukemias”的研究论文。该研究通过多学科协作,首次发现去SUMO化兼去泛素化酶DESI2是维持JAK2-V617F稳定性的“关键守护者”。基于此靶点,研究团队成功开发出能精准诱导突变蛋白降解的小分子抑制剂WWQ-03-012,在临床前研究中展现出显著疗效。该成果不仅为这类难治性血液肿瘤提供了从“抑制”到“清除”的全新治疗范式,也为克服靶向治疗耐药开辟了切实可行的新路径。

为了寻找能够特异性调控突变型JAK2的关键分子,研究团队采用免疫共沉淀联合质谱分析技术。通过对比表达野生型和JAK2-V617F突变蛋白的细胞,发现DESI2在突变型JAK2的互作蛋白中特异性富集。进一步的检测显示,DESI2在JAK2-V617F阳性的白血病细胞系及患者原代样本中均高表达,表明其与突变疾病的潜在关联。
为确证DESI2与JAK2-V617F结合的特异性,研究团队设计了多重正交实验。在等基因背景的Ba/F3细胞中,DESI2仅与突变型JAK2结合。同一细胞系中外源表达相同水平的野生型和突变型JAK2,也验证了DESI2的选择性结合。免疫荧光分析显示,DESI2与JAK2-V617F在细胞质中的共定位信号更强。通过AlphaFold 3结构模拟发现,突变导致的构象变化使DESI2与JAK2-V617F形成了多个氢键网络,从空间上解释了结合的特异性。
针对DESI2的作用机制研究揭示,DESI2通过其去SUMO化酶活性,移除JAK2蛋白K962位点的SUMO化修饰,从而抑制该位点的泛素化,阻断蛋白酶体对JAK2-V617F的降解。AlphaFold 3模型显示,DESI2的催化中心精准定位在JAK2-V617F的K962位点附近,阐明了其高效特异的催化机制。遗传学回补实验进一步证实,DESI2对JAK2-V617F的稳定作用严格依赖其酶活性,且仅针对突变型蛋白。
在验证DESI2作为治疗靶点的潜力方面,细胞实验表明敲低DESI2能够选择性降解JAK2-V617F蛋白、抑制下游致癌信号并诱导突变细胞凋亡。在JAK2-V617F阳性白血病异种移植模型中,DESI2敲除显著抑制了肿瘤生长、降低了骨髓白血病负荷并延长了小鼠生存期,而在野生型模型中无此效应。在JAK2-V617F基因敲入MPN模型中,敲低DESI2可有效缓解疾病表征、改善血象并延长生存期,且不影响正常造血功能。
基于对DESI2作用机制的深入理解,研究团队通过高通量筛选发现了苗头化合物F-70。通过基于结构的药物设计,在苯并噻唑母核的C8位引入芳香环结构,增强了与DESI2的结合能力,最终获得了优化化合物WWQ-03-012。该化合物的抑制活性较先导化合物提升了数百倍,并在化学蛋白组学分析中表现出高度选择性。
在全面评估环节,WWQ-03-012在细胞层面能够剂量依赖性地降解JAK2-V617F蛋白,选择性抑制突变细胞增殖,而对野生型细胞或正常造血细胞影响甚微。在临床来源的原代细胞中,该化合物同样表现出对JAK2-V617F阳性细胞的选择性抑制。动物实验进一步证实,WWQ-03-012可显著抑制JAK2-V617F白血病细胞在小鼠体内的生长,改善MPN模型小鼠的血象指标,且在健康野生型小鼠中表现出良好的耐受性。尤为重要的是,该化合物能够有效抑制鲁索替尼耐药细胞株的生长,并与鲁索替尼联用展现协同效应,为临床克服耐药提供了有力依据。
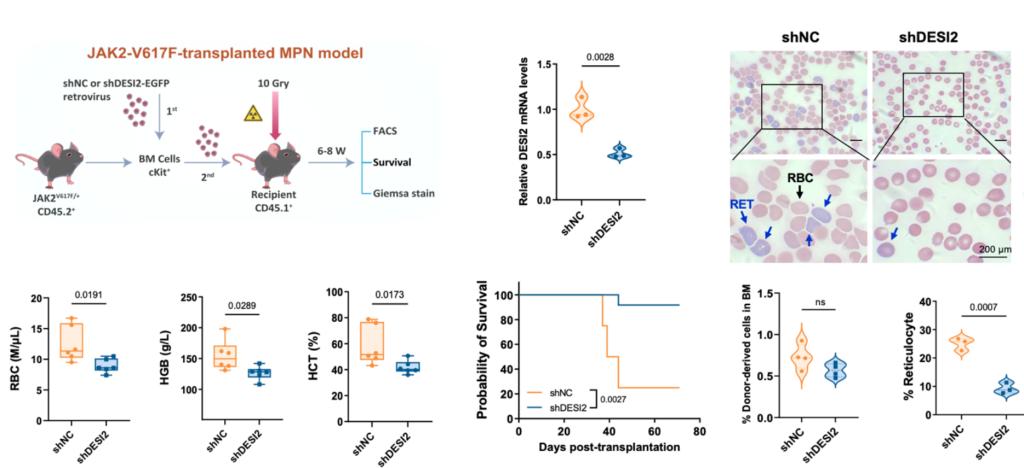
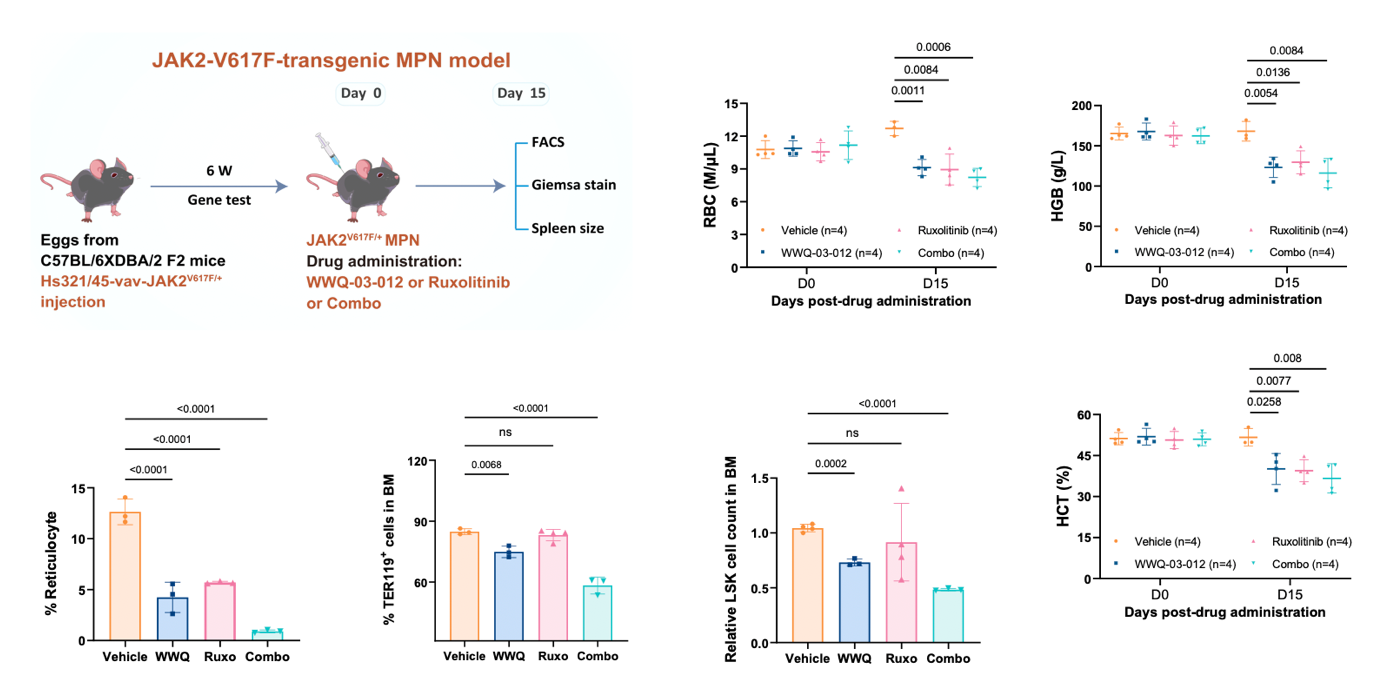
该研究不仅鉴定出DESI2是维持JAK2-V617F致癌稳定性的关键上游调控因子,更成功开发出首个靶向其酶活性的高选择性小分子降解剂WWQ-03-012。这项工作实现了从“靶点发现”到“药物创制”的完整闭环,具有多重重要意义:1) 提供了颠覆传统激酶抑制的新范式:与鲁索替尼等ATP竞争性抑制剂不同,WWQ-03-012通过诱导致癌蛋白的彻底降解,从源头上消除其功能,有望实现更深入、更持久的病理阻断,为克服因靶点突变或信号代偿导致的临床耐药提供了全新解决方案。2)揭示了蛋白质翻译后修饰网络的精密调控:研究阐明了DESI2通过协调K962位点的“去SUMO化-去泛素化”双重修饰来稳定JAK2-V617F的精细机制。这一发现加深了我们对SUMO化与泛素化交叉对话(crosstalk)在肿瘤发生中作用的理解,也为干预其他依赖类似修饰网络的疾病提供了理论参考。3)展现了转化医学的强力驱动:从临床问题(JAK2突变白血病耐药)出发,利用蛋白质组学筛选发现新靶点,再通过结构生物学指导的理性药物设计得到候选化合物,并在患者来源样本和多种动物模型中验证疗效,整个研究链条紧密围绕临床转化需求,显著提升了成果的临床应用潜力。
基于现有突破,团队已通过结构优化研发出活性与选择性显著优于WWQ-03-012的新一代候选化合物WWQ-06-018。同时,DESI2与抑制剂及JAK2-V617F相互作用界面的高分辨率结构解析工作也在推进,将为临床候选药物的理性设计提供关键蓝图。
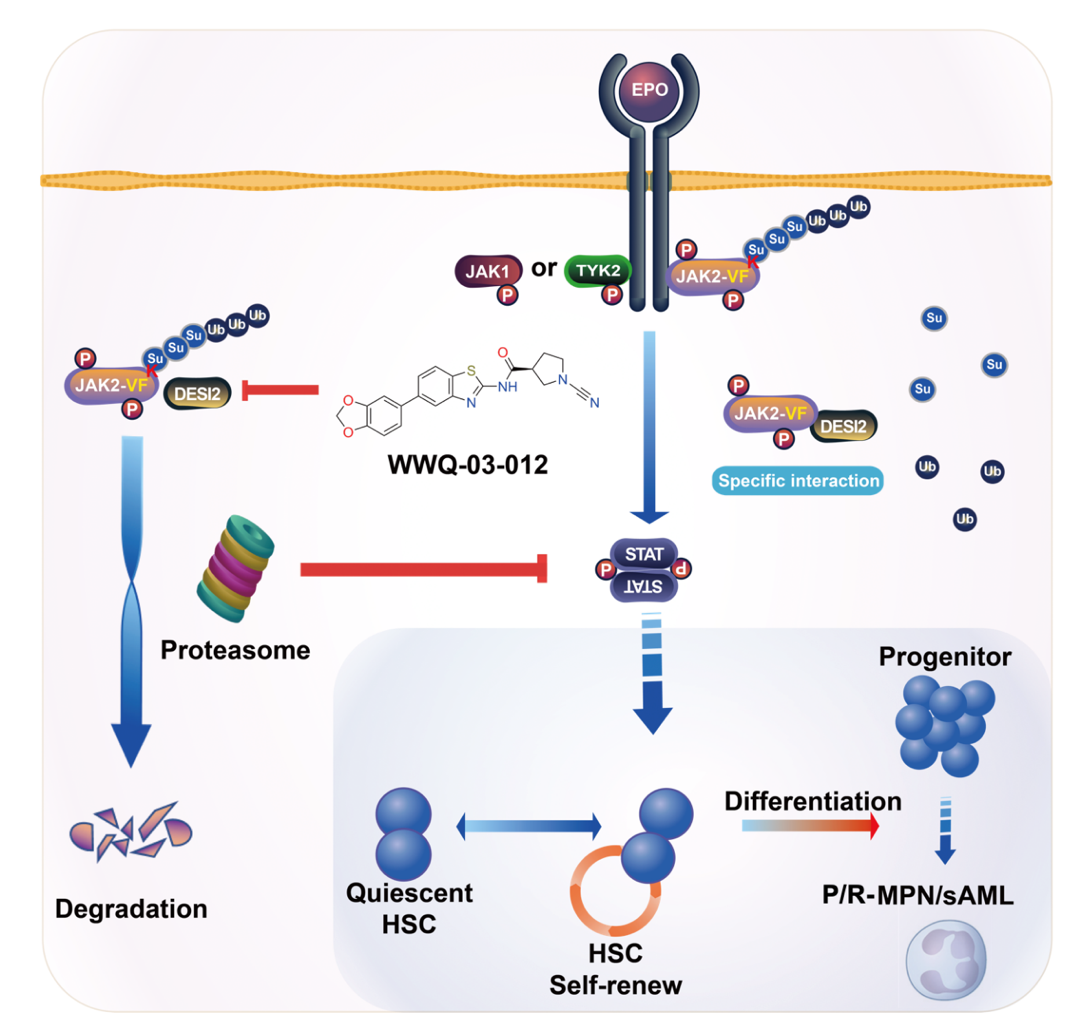
总之,这项研究不仅为JAK2突变型血液恶性肿瘤带来了新的希望之光,更为克服临床耐药这一棘手难题提供了全新的解决路径。DESI2作为一个极具潜力的新靶点,连同其高效抑制剂WWQ-03-012及后续更优的化合物,有望在未来成为临床治疗武器库中的重要一员,通过诱导突变蛋白降解这一新机制,为耐药患者提供更有效的治疗选择,并推动肿瘤靶向治疗迈向“降解时代”。
杨静教授、蒋白山教授、梁爱斌教授及赵保兵教授为论文共同通讯作者。同济大学博士后梅沪生、武汉大学博士后文武强、同济大学附属同济医院张文君主任、山东大学博士生张淑敬、中国医学科学院血液病研究所孙婷医师为论文共同第一作者。该研究获国家自然科学基金、国家重点研发计划等项目支持。
论文链接:https://advanced.onlinelibrary.wiley.com/doi/epdf/10.1002/advs.202515127



